
 购物车件
购物车件
 English
English
JACS:南开大学汤平平课题组实现钴催化的环氧化物三氟甲氧基化
2018-12-26
导读
近日,南开大学汤平平课题组在J. Am. Chem. Soc.首次报道了温和条件下钴催化的环氧化物与三氟甲基芳基磺酸盐(TFMS)的开环反应(Figure 1) (doi: 10.1021/jacs.8b10298)。
环氧化物的亲核开环是通过单步反应获得有机分子双官能团的重要方法,主要亲核试剂包括胺、硫醇、羟基、苯酚、羧酸、叠氮化物、氰化物、卤素等;相比之下,由于三氟甲氧基(OCF3)阴离子的弱亲核性和不稳定性,限制了其用作环氧化物开环反应中的亲核试剂。此外,使用高压(4 MP)的有毒氟光气作为三氟甲氧基阴离子(-OCF3)的来源增加了操作的风险和难度。因此,三氟甲氧基阴离子对环氧化物的亲核开环极具挑战性。
最近,汤平平课题组报道了TFMS作为三氟甲氧基化试剂的一系列应用,其三氟甲氧基阴离子-OCF3可由AgF、CsF原位生成(Figure 1a)。作者猜测是否有可能通过TFMS的三氟甲氧基阴离子实现环氧化物的亲核开环。其面临主要问题包括:a)由于氟化物阴离子可能发生环氧化物的亲核开环,所以氟化物盐不适合激活TFMS以产生-OCF3;b)如何稳定三氟甲氧基阴离子。对于以上问题,作者假设催化剂SalenCoX(Figure 1c)的轴向阴离子X可与TFMS反应产生-OCF3,并对环氧化物(1)开环得到中间体A,其与从-OCF3原位产生的氟光气反应形成中间体B,然后经后处理得到所需的邻位三氟甲氧基氢化物(3);从-OCF3分解的氟化物进一步用于在反应过程中用TFMS产生-OCF3(Figure 1b)。
(图片来源:J. Am. Chem. Soc.)
作者以2,3-环氧-1,2,3,4-四氢化萘(1a)作为模型底物对反应条件进行了优化(Table 1)。首先,筛选了SalenCoX的不同轴向阴离子X。与其它Lewis酸相比,当使用10% mol SalenCoOTs作为催化剂时,以17%产率得到产物3a;吸电子轴向阴离子X如PhCO2-, CF3CO2-和DNP催化活性最佳,而DNP的催化产率最高(73%)。相反,亲核性差的阴离子如BF4-和SbF6-抑制了产物的形成。轴向阴离子DNP的作用不仅易于与金属中心解离以活化环氧化物,而且还可以与TFMS反应生成-OCF3。Langlois及其同事报道n-Bu4N+可稳定-OCF3,因此当使用10% mol n-Bu4N+DNP-得到最高产率(88%),而进一步增加铵盐的量对反应则具有负面影响。当使用3.0 eq.的TFMS时,产率进一步提高至97%。通过对照反应发现,无钴催化剂时没有产生三氟甲氧基化产物。在对反应条件进行广泛优化后,最终发现1a于室温氮气保护情况下,在乙腈中使用10% mol催化剂I,10% mol n-Bu4N+DNP-, 3 eq TFMS (2)进行反应时,可以97%产率得到3a。
(图片来源:J. Am. Chem. Soc.)
在优化的条件下,作者研究了内消旋-环氧化物与TFMS开环反应的适用范围(Table 2)。研究发现,五元、六元、七元环底物可以45%~95%的产率转化为预期的邻位三氟甲氧基氢化物,产率范围为;非环状底物1e和1f也可以高产率转化为所需产物(3e,3f)。此外,该转化反应可以耐受底物中一些官能团(如酯,醚和酰胺等)。另外,通过氟化物进行环氧化物开环,可以得到少于5%的氟化副产物。
(图片来源:J. Am. Chem. Soc.)
除了内消旋-环氧化物外,作者还研究了外消旋环氧化物(Table 3)。研究发现,末端环氧化物更容易转化为邻位三氟甲氧基氢化物(3m-3r);并且醚、腈和卤素等基团均耐受。此外,作者发现催化体系在底物1m-1p的开环中具有优异的区域选择性,仅生成仲醇3m至3p;底物1q和1r反应中具有高区域选择性,并且发现少于2%的伯醇副产物。受此启发,作者认为该催化体系可以在开环过程中保留次甲基碳的立体化学。因此,作者利用手性纯底物(S)-1m以88%的产率得到产物3m(ee>99%),表明催化剂I主要在(S)-1m的亚甲基碳上诱导开环,并保持次甲基碳的立体化学。然而,当用苯乙烯环氧化物1s作底物时,由于空间和电性因素与亲核进攻冲突,产生较低的区域选择性。不对称内式环氧-烷烃1t的转化表明空间因素对开环位置具有很大影响,-OCF3更有利于进攻环氧中位阻较小的碳得到产物3t作为主要产物。除区域选择性外,催化剂I在具有两个不同环氧基的底物1u开环中也显示出了优异的化学选择性,并且由于左环氧基具有空间位阻,开环仅发生在末端环氧基处。
(图片来源:J. Am. Chem. Soc.)
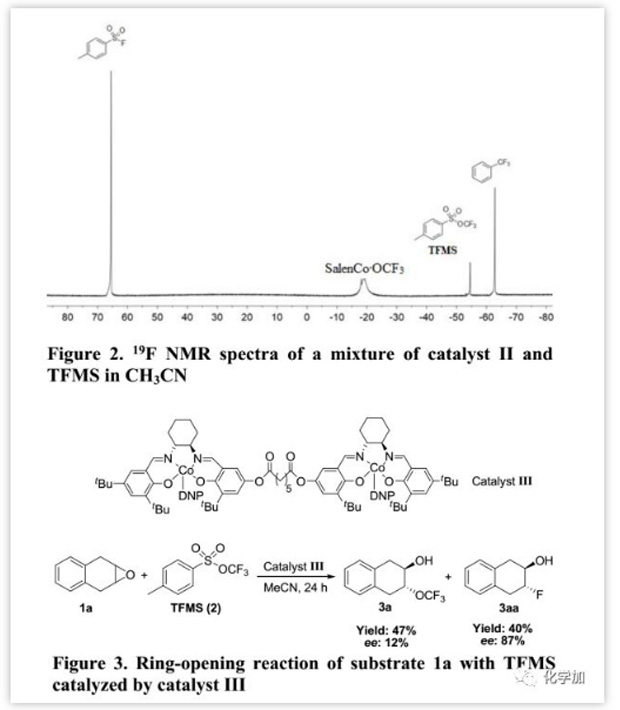
最后,作者还研究了通过三氟甲氧基化阴离子对环氧化物的不对称开环。起初,用手性纯催化剂II进行内消旋-环氧化物(1a)的去对称化,仅以95%收率和6%的ee值得到产物3a,并且外消旋-环氧化物(1m)的动力学拆分产物3m产率为80%,ee值为13%。为解释此现象,作者进行了初步研究以了解其反应机制。Jacobsen报道金属salen复合物不仅作为Lewis酸活化环氧化物,还通过双金属配合作为亲核试剂,这是实现环氧化物高对映选择性开环的关键点。因此,作者通过19F NMR监测在乙腈中催化剂II和TFMS的混合物以表明SalenCoOCF3的存在(约-18.8 ppm,Figure 2)。此外,关于催化剂II的动力学研究揭示了速率常数(k)与[催化剂II]之间的线性相关性,反映了对催化剂的二级依赖性;通过利用Burés开发的可变时间归一化分析图形方法分析数据也得出了相同的结论。这些观察结果提供的确凿证据,表明在环氧化物与TFMS的开环反应中存在双金属配合。Jacobsen进一步提出了两种限制几何形状的对映体确定过渡状态:“头对头”和“头对尾”。其中,只有“头对尾”的几何形状才能诱导环氧化物的不对称开环,这可以通过二聚体salen-铬络合物(类似于Figure 3中的催化剂III)证实,其可以通过分子内双金属途径模拟“头到尾”的几何形状。然而,当用手性催化剂III进行1a与TFMS开环时,除产生三氟甲氧基化产物3a(47%,19F NMR产率)外,还存在氟化产物3aa(40%,19F NMR产率);这表明氟化产物3aa是通过分子内双金属途径(“头对尾”)产生的,而三氟甲氧基化产物3a不是。因此,催化剂II的一个钴中心很难诱导-OCF3通过“头到尾”几何形状接近由另一个钴中心活化的环氧化物。阻碍屏障可能是由CF3和活化的环氧化物或salen配体之间的空间排斥引起的,并且这也可以解释为何催化剂II在TFMS进行环氧化物开环中立体控制性丧失。
(图片来源:J. Am. Chem. Soc.)
小结:南开大学汤平平课题组开发了一种钴催化的通过三氟甲基芳基磺酸盐(TFMS)对环氧化物进行亲核开环产生三氟甲氧基化产物的反应。该反应可以在温和条件下直接构建多种邻位三氟甲氧基羟基化合物,并且具有广泛的官能团耐受性,另外,该方法还可以将末端环氧化物转化为具有良好化学和区域选择性的目标产物,在含三氟甲氧基的活性化合物合成中具有重要应用价值。

